HLB가 미국 식품의약국(FDA)에 항암 신약 '리보세라닙'의 신약 판매 허가 신청서(NDA)를 제출했다. 리보세라닙을 개발하기 시작한 지 16년, HLB가 글로벌 임상을 시작한 지 12년 만이다. 일반적으로 NDA를 제출한 뒤 FDA 최종 품목허가 여부가 결정되기까지는 10개월가량이 소요된다.
17일 제약바이오 업계에 따르면 HLB는 지난 16일(미국 시각) FDA에 간암 1차 치료제를 적응증으로 리보세라닙의 NDA를 제출했다.
리보세라닙은 암 형성에 관여하는 신생 혈관 생성을 억제하는 원리의 2세대 표적항암제다. 암젠의 수석연구원이자 주요 투자자로도 활동했던 폴 첸 미국 어드벤첸 연구소 대표가 지난 2003년부터 개발하기 시작한 후보물질이다. 이후 HLB가 리보세라닙의 글로벌 특허권을 확보, 2011년부터 위암·간세포암·대장암·선양낭성암 등 여러 적응증으로 다국가 임상을 진행해 왔다.
리보세라닙은 중국 시장에서는 이미 '아이탄'이라는 이름으로 상용화에 성공했다. 리보세라닙의 중국 판권을 보유한 중국 항서제약이 지난 2014년 현지 규제당국으로부터 위암 3차 치료제로 리보세라닙의 품목허가를 획득한 바 있다. 이어 2020년 12월 간세포암 2차 치료제로도 승인받아 시판 중이다. 참고로 리보세라닙의 한국 판권은 HLB 자회사 HLB생명과학이, 한국·중국을 제외한 나머지 국가 판권은 HLB 미국 자회사인 엘레바가 보유하고 있다.
그러나 HLB는 리보세라닙의 미국 시장 진출에서 매번 고배를 마셨다. 회사는 2019년 리보세라닙의 위암 3·4차 치료제 다국가 임상에 실패한 경험이 있다. 위암 2차 이상 표준 치료에 실패한 말기 환자를 대상으로 한 임상3상에서 1차 유효성 평가지표를 달성하지 못했다. 당시 회사는 세부 데이터를 확인했을 때 임상적 유의성이 있다고 판단해 위암 3·4차 치료제로 NDA를 진행하겠다고 했지만 이제껏 지연됐다. 또 지난해 선양낭성암을 적응증으로 연내 NDA를 제출하겠다는 계획을 내놨지만, 이 역시 FDA의 추가 자료 요청으로 미뤄졌다.
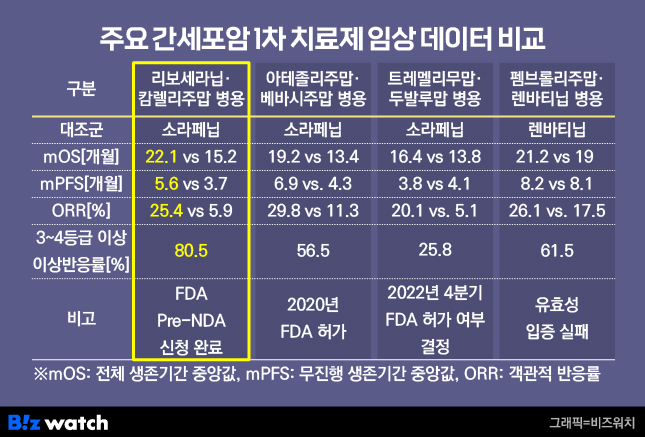
이번 NDA는 간세포암 1차 치료제 임상 결과를 기반으로 제출했다. 해당 임상은 리보세라닙과 항서제약의 면역항암제 '캄렐리주맙'을 함께 투여하는 병용요법으로 진행했다. 13개국 543명의 환자를 대상으로 진행한 다국가 임상3상(CARES 310) 결과, 1차 유효성 평가지표인 전체 생존기간(OS)*과 무진행생존기간(PFS)**이 대조군인 '소라페닙'(제품명 넥사바)보다 길게 나타났다. 특히 리보세라닙의 전체 생존기간 중앙값(mOS) 22.1개월은 간암 치료제 중 가장 긴 생존기간으로, 높은 치료 효능을 입증했다는 평가를 받는다.
*전체 생존기간(OS): 암 진행 여부와 상관없이 환자가 생존한 기간
**무진행생존기간(PFS): 암이 일정 크기 이상으로 커지지 않고 생존한 기간
다만 3등급 이상 부작용 발생률이 높게 나타난 점은 FDA 허가의 걸림돌로 꼽힌다. 임상3상에서 리보세라닙과 캄렐리주맙을 병용 투여받은 환자의 80.5%에서 3~4등급 이상의 이상반응이 발생했다. 간암 치료에선 OS 개선 외에도 독성 관리가 중요하게 여겨진다. 이에 대해 HLB 측은 "3~4등급 이상의 부작용 발생률이 80%가량으로 높아 보이지만 모두 관리 가능한 수준"이라면서 "실제 절반 수준이 고혈압으로 이는 고혈압 약으로 충분히 관리할 수 있다"고 답한 바 있다.
통상 NDA를 제출한 뒤 FDA 최종 품목허가 여부가 결정되기까지는 10개월가량이 소요된다. 우선 FDA는 NDA가 접수되면 60일 동안 서류 심사를 진행할지 결정한다. 기업이 신약 품목허가 신청을 위해 준비한 자료가 검토하기 적정한지 확인하는 절차다. 서류심사 등록 절차가 끝나면 FDA는 14일 내로 NDA 등록 승인 여부를 통보한다. 이후 신약 후보물질에 대한 정식 심사, 생산시설 심사 등을 거친다.
HLB 측은 "FDA가 리보세라닙 임상3상 결과를 바탕으로 신약허가 신청 전 미팅(pre-NDA)을 진행한 후 리보세라닙 병용요법에 대한 NDA 제출에 '문제가 없다'고 밝혔다"면서 "현재까지 신생혈관억제 기전의 TKI 항암제와 면역항암제의 병용요법으로 허가받은 간암 1차 치료제가 없다는 점도 두 약물의 허가 기대감을 높이는 요인"이라며 FDA 품목허가에 대한 자신감을 드러냈다. 회사는 리보세라닙 품목허가 후 빠른 판매를 위해 생산 및 판매·유통 등 상업화 준비에도 나선 상태다.
리보세라닙이 성공적으로 FDA 품목허가를 받게 되면 연 매출 1조원을 거두는 블록버스터 항암제로 성장할 것이라는 전망도 나온다. 간암은 전체 암 중 6번째로 많이 발병하는 질환인데, 뚜렷한 치료 효과를 내는 치료제는 없는 상황이다. 여기에 리보세라닙은 먹는(경구용) 항암제로, 환자 편의성과 가격 측면에서 경쟁력을 갖췄다는 게 HLB 측의 설명이다. 기존 간세포암 1차 치료제로 FDA 허가를 받은 로슈의 티센트릭과 아바스틴 병용요법의 경우 두 약물이 모두 주사제다. 해당 요법의 연간 약가는 30만달러(약 4억원)에 달한다.
정세호 엘레바 대표는 "이번 NDA 제출로 당사가 중요한 이정표를 세웠다"면서 "많은 임상의사들로부터 임상 결과에 대해 좋은 평가를 받아온 만큼 무난히 신약 허가를 받을 수 있다고 본다"고 했다.